Linfangioleiomiomatosis
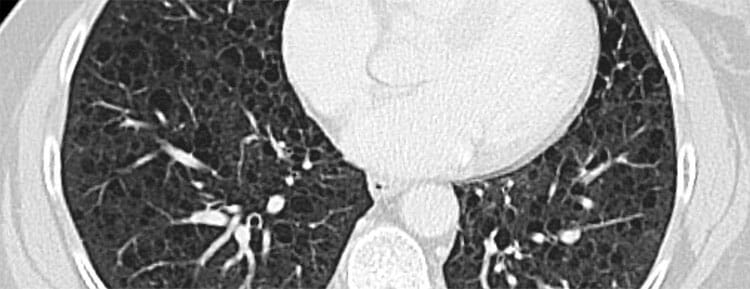
Tabla de contenidos
- 1.¿Qué es la linfangioleiomiomatosis?
- 2.Causas de la linfangioleiomiomatosis
- Factores hormonales
- Mecanismo patológico
- 3.Síntomas de la linfangioleiomiomatosis
- Síntomas pulmonares
- Síntomas extrapulmonares
- 4.Diagnóstico de la linfangioleiomiomatosis
- Pruebas de imagen
- Pruebas de laboratorio
- Pruebas funcionales respiratorias
- Biopsia
- 5.Tratamiento de la linfangioleiomiomatosis
- Sirolimus (rapamicina)
- Tratamiento hormonal
- Tratamiento de las complicaciones
- Trasplante pulmonar
- Oxigenoterapia y rehabilitación pulmonar
- 6.Pronóstico
- 7.¿Cómo puedo evitarla?
- 8.Cuándo acudir al médico
- 9.Vivir con linfangioleiomiomatosis
- 10.Referencias
La linfangioleiomiomatosis (LAM) es una enfermedad pulmonar rara y progresiva, perteneciente a la categoría de enfermedades pulmonares intersticiales difusas. Bajo esta denominación se agrupan procesos de distinto origen en los que se afectan las paredes de los alvéolos y el tejido perialveolar. La LAM no es una enfermedad maligna y no está causada por agentes infecciosos definidos, aunque las células LAM presentan un comportamiento similar al de las células tumorales de bajo grado, ya que proliferan de forma anómala y pueden diseminarse.
El comienzo es insidioso, no agudo, y la enfermedad suele tener un curso crónico y progresivo. La respuesta patológica inicial consiste en la proliferación de células musculares lisas inmaduras (denominadas células LAM) en las paredes de los alvéolos, las vías aéreas, los vasos sanguíneos y los vasos linfáticos del pulmón. Esta proliferación causa obstrucción de las estructuras afectadas y, con el tiempo, da lugar a la formación de quistes pulmonares que van destruyendo progresivamente el parénquima pulmonar.
¿Qué es la linfangioleiomiomatosis?
La LAM es una enfermedad que se caracteriza por la proliferación descontrolada de un tipo especial de células musculares lisas anómalas (células LAM) en los pulmones y, en algunos casos, en otros órganos como los riñones, los ganglios linfáticos y el útero. Estas células expresan receptores de estrógenos y progesterona, lo que explica la estrecha relación de la enfermedad con las hormonas sexuales femeninas.
Se estima que la LAM afecta a entre 3 y 8 mujeres por cada millón de habitantes, lo que la convierte en una enfermedad rara o minoritaria. Existen dos formas de la enfermedad:
- LAM esporádica: aparece de forma aislada, sin asociación con otras enfermedades genéticas. Es la forma más frecuente.
- LAM asociada al complejo esclerosis tuberosa (CET): la esclerosis tuberosa es una enfermedad genética que causa tumores benignos en múltiples órganos. Aproximadamente un 30-40 % de las mujeres con CET desarrollan LAM pulmonar.
Causas de la linfangioleiomiomatosis
La causa de la LAM se ha identificado en las mutaciones de los genes TSC1 o TSC2 (genes del complejo de la esclerosis tuberosa). Estos genes codifican las proteínas hamartina y tuberina, respectivamente, que forman un complejo encargado de regular la vía de señalización celular mTOR (mammalian target of rapamycin). Cuando estos genes están mutados, la vía mTOR se activa de forma excesiva, lo que provoca una proliferación celular descontrolada.
- En la LAM esporádica, las mutaciones de TSC2 se producen de forma somática (adquirida) exclusivamente en las células LAM.
- En la LAM asociada a CET, las mutaciones de TSC1 o TSC2 son germinales (heredadas) y afectan a todas las células del organismo.
Factores hormonales
Afecta casi exclusivamente a mujeres en edad fértil (generalmente entre los 20 y los 40 años). El hecho de que la enfermedad empeore durante el embarazo, el posparto y después de tratamiento con estrógenos parece confirmar que existe una asociación directa entre este trastorno y la secreción de estrógenos (hormonas sexuales femeninas). Las células LAM expresan receptores de estrógenos y progesterona, lo que sugiere que estas hormonas favorecen su proliferación.
Mecanismo patológico
Las células musculares lisas inmaduras pueden proliferar en el tejido pulmonar alrededor de las estructuras bronquiales, vasculares y linfáticas y en su interior, provocando obstrucción local y creando lesiones constrictivas que, con el tiempo, se transforman en quistes de paredes delgadas. También se pueden afectar vasos y ganglios linfáticos de otros órganos, incluyendo el mediastino, el retroperitoneo y la pelvis. En los riñones pueden formarse angiomiolipomas, tumores benignos compuestos por vasos sanguíneos, músculo liso y tejido adiposo.
Síntomas de la linfangioleiomiomatosis
La LAM suele manifestarse de forma gradual. Los síntomas pueden ser muy variables y, en las fases iniciales, a menudo se confunden con otras enfermedades respiratorias como el asma o la EPOC. Las manifestaciones clínicas más frecuentes son:
Síntomas pulmonares
- Disnea progresiva: dificultad para respirar que empeora con el esfuerzo. Es el síntoma más frecuente y suele ser el motivo de consulta principal.
- Tos seca: persistente y, en ocasiones, acompañada de sibilancias.
- Neumotórax espontáneo: colapso parcial o total del pulmón por la rotura de quistes subpleurales. Es la forma de presentación en un 40-50 % de las pacientes y puede ser recurrente.
- Hemoptisis: expulsión de sangre con la tos, debida a la obstrucción y rotura de vasos pulmonares. Suele ser leve.
Síntomas extrapulmonares
- Derrame quiloso (quilotórax): acumulación de líquido linfático (quilo) en la cavidad pleural, debido a la obstrucción de los vasos linfáticos. Puede causar disnea y dolor torácico.
- Ascitis quilosa: acumulación de quilo en la cavidad abdominal, menos frecuente.
- Angiomiolipomas renales: presentes en un 30-40 % de las pacientes con LAM esporádica y en la mayoría de las pacientes con LAM asociada a CET. Suelen ser asintomáticos, pero pueden causar dolor lumbar o hemorragia si alcanzan un tamaño considerable.
- Linfangioleiomiomas abdominales o pélvicos: masas de tejido linfático y muscular liso que pueden causar dolor o sensación de masa abdominal.
Diagnóstico de la linfangioleiomiomatosis
Pruebas de imagen
- Radiografía de tórax: puede mostrar imágenes reticulonodulares (áreas que asemejan redes que se cruzan y zonas de aspecto nodular) y pequeñas áreas quísticas, aunque en fases iniciales puede ser normal.
- Tomografía computarizada de alta resolución (TCAR): es la prueba de imagen más importante para el diagnóstico. Muestra quistes pulmonares bilaterales de paredes delgadas, distribuidos de forma difusa y homogénea por ambos pulmones. Este patrón es muy característico y, en el contexto clínico adecuado, puede ser suficiente para establecer el diagnóstico.
- TAC abdominal: para detectar angiomiolipomas renales y linfangioleiomiomas abdominales.
- Resonancia magnética: útil para la evaluación de lesiones abdominales y pélvicas.
Pruebas de laboratorio
- VEGF-D (factor de crecimiento del endotelio vascular tipo D): los niveles séricos elevados de VEGF-D (por encima de 800 pg/mL) tienen una alta especificidad para el diagnóstico de LAM y pueden evitar la necesidad de biopsia en muchos casos. También es útil para monitorizar la respuesta al tratamiento.
Pruebas funcionales respiratorias
- Espirometría: suele mostrar un patrón obstructivo (disminución del FEV1) o mixto.
- Difusión de monóxido de carbono (DLCO): suele estar reducida y es un indicador sensible de la gravedad de la enfermedad.
- Prueba de esfuerzo cardiopulmonar: valora la capacidad de ejercicio y la desaturación de oxígeno con el esfuerzo.
Biopsia
La clínica y los hallazgos radiológicos, junto con la determinación de VEGF-D, pueden ser suficientes para el diagnóstico en muchos casos. Sin embargo, cuando existe duda diagnóstica, el diagnóstico se establece de forma definitiva con la biopsia transbronquial o pulmonar (quirúrgica, mediante videotoracoscopia). Las células LAM presentan una tinción positiva para actina de músculo liso, HMB-45 (un marcador melanocítico) y receptores de estrógenos y progesterona, lo que las distingue de otras patologías.
Tratamiento de la linfangioleiomiomatosis
El tratamiento de la LAM ha experimentado avances significativos en las últimas décadas gracias al mejor conocimiento de los mecanismos moleculares de la enfermedad.
Sirolimus (rapamicina)
El sirolimus es actualmente el tratamiento de primera línea para la LAM. Es un inhibidor de la vía mTOR que actúa directamente sobre el mecanismo molecular que causa la proliferación de las células LAM. Los ensayos clínicos (especialmente el estudio MILES, publicado en The New England Journal of Medicine en 2011) demostraron que el sirolimus:
- Estabiliza o mejora la función pulmonar (FEV1 y capacidad vital forzada).
- Reduce el tamaño de los angiomiolipomas renales.
- Puede reducir los derrames quilosos.
El sirolimus se indica habitualmente cuando la función pulmonar está deteriorada o en descenso progresivo (FEV1 < 70 % del predicho o descenso significativo), cuando hay derrames quilosos recurrentes o cuando los angiomiolipomas renales son grandes o sintomáticos.
Tratamiento hormonal
El tratamiento con acetato de medroxiprogesterona (progesterona) fue utilizado históricamente, pero los estudios posteriores no han demostrado su eficacia de forma consistente. Actualmente no se recomienda como tratamiento de primera línea.
Se recomienda evitar los tratamientos con estrógenos (anticonceptivos orales con estrógenos, terapia hormonal sustitutiva) ya que pueden empeorar la enfermedad.
Tratamiento de las complicaciones
- Neumotórax: puede requerir drenaje torácico. Dado que la recurrencia es muy alta (hasta el 70 %), se recomienda la pleurodesis química o quirúrgica (unión de las capas pleurales para evitar nuevos neumotórax) tras el primer episodio. La pleurodesis no es una contraindicación para un futuro trasplante pulmonar.
- Derrame quiloso: puede tratarse con drenaje, restricción dietética de grasas (dieta con triglicéridos de cadena media), sirolimus o, en casos refractarios, cirugía.
- Angiomiolipomas renales: los de gran tamaño (> 4 cm) o sintomáticos pueden tratarse con sirolimus, embolización selectiva o, raramente, cirugía.
Trasplante pulmonar
En las fases avanzadas de la enfermedad, cuando la insuficiencia respiratoria es grave y no responde a otros tratamientos, el trasplante pulmonar es una alternativa terapéutica. La supervivencia tras el trasplante en pacientes con LAM es similar a la de otras indicaciones.
Oxigenoterapia y rehabilitación pulmonar
La oxigenoterapia domiciliaria se indica cuando hay hipoxemia crónica. Los programas de rehabilitación pulmonar pueden mejorar la capacidad de ejercicio y la calidad de vida.
Pronóstico
El pronóstico de la LAM ha mejorado significativamente con la introducción del sirolimus. La supervivencia media desde el diagnóstico supera actualmente los 20-30 años en la mayoría de las series. La evolución es variable: algunas pacientes presentan una enfermedad estable durante años, mientras que otras experimentan un deterioro progresivo de la función pulmonar. Los factores que se asocian a un peor pronóstico incluyen:
- Función pulmonar inicial más deteriorada.
- Mayor velocidad de descenso del FEV1.
- Presencia de VEGF-D muy elevado.
- Presentación con neumotórax recurrente.
¿Cómo puedo evitarla?
Al tratarse de una enfermedad causada por mutaciones genéticas, no es posible realizar ninguna medida que prevenga su aparición. Sin embargo, el diagnóstico precoz es fundamental para iniciar el tratamiento con sirolimus en el momento adecuado y mejorar el pronóstico.
Cuándo acudir al médico
Si presentas alguno de los siguientes síntomas, especialmente si eres mujer en edad fértil, debes acudir a tu médico para una evaluación:
- Disnea de esfuerzo progresiva sin causa aparente.
- Neumotórax espontáneo (dolor torácico brusco que aumenta con la inspiración profunda y se acompaña de sensación de falta de aire), especialmente si es recurrente.
- Hemoptisis (expectoración de sangre roja en cantidad variable con los accesos de tos).
- Tos persistente que no mejora con tratamientos habituales.
- Si te han diagnosticado esclerosis tuberosa, es importante realizar cribado periódico con TCAR para detectar LAM precozmente.
El médico debe realizar el diagnóstico diferencial con otros procesos pulmonares con manifestaciones similares, como la histiocitosis pulmonar de células de Langerhans, el síndrome de Birt-Hogg-Dubé y el enfisema.
Vivir con linfangioleiomiomatosis
El diagnóstico de una enfermedad rara como la LAM puede generar incertidumbre y preocupación. Algunas recomendaciones para las pacientes:
- Seguimiento regular por un neumólogo especializado en enfermedades pulmonares intersticiales.
- Vacunación: mantener actualizado el calendario vacunal, incluyendo la vacuna antigripal anual y la vacuna antineumocócica.
- Evitar el tabaco: fumar empeora cualquier enfermedad pulmonar.
- Ejercicio físico adaptado: la actividad física regular, dentro de las posibilidades de cada paciente, mejora la calidad de vida.
- Precaución con los viajes en avión: los cambios de presión en la cabina pueden aumentar el riesgo de neumotórax. Consulta con tu neumólogo antes de viajar.
- Anticoncepción sin estrógenos: se deben evitar los anticonceptivos que contengan estrógenos. Los métodos recomendados incluyen dispositivos intrauterinos (DIU) con levonorgestrel, implantes de progestágeno o métodos de barrera.
- Asociaciones de pacientes: conectar con otras pacientes a través de asociaciones como la Asociación Española de Linfangioleiomiomatosis (AELAM) o la LAM Foundation puede proporcionar apoyo emocional, información actualizada y acceso a recursos.
Referencias
- McCormack, F. X., et al. (2011). Efficacy and safety of sirolimus in lymphangioleiomyomatosis. The New England Journal of Medicine, 364(17), 1595-1606.
- Johnson, S. R., et al. (2010). European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. European Respiratory Journal, 35(1), 14-26.
- Gupta, N., et al. (2017). Lymphangioleiomyomatosis: Diagnosis and Management. American Journal of Respiratory and Critical Care Medicine, 196(6), 690-699.
- Orphanet. Linfangioleiomiomatosis. Disponible en: https://www.orpha.net
- MedlinePlus. Lymphangioleiomyomatosis. Biblioteca Nacional de Medicina de EE. UU. Disponible en: https://medlineplus.gov/genetics/condition/lymphangioleiomyomatosis/
- Sociedad Española de Neumología y Cirugía Torácica (SEPAR). Enfermedades pulmonares intersticiales difusas. Disponible en: https://www.separ.es

Escrito por
Gabriel GinerEditor
Fundador y editor de eSalud. Apasionado de la salud digital y la divulgación sanitaria, dirige el proyecto editorial desde sus inicios con el compromiso de acercar la información de salud a todos los lectores.